Contextualize your Data¶
The generation of a new design population can provide a lot of data depending in the number of filters and metrics used to evaluate them. Still, although these scores allow for the sorting and selection of decoys of interest, it does not always provide a comprehensive analysis of that population with regard to the general population of known protein structures.
The rstoolbox provides some helper functions and datasets in order to properly assess this issue.
Pre-calculated datasets¶
There is a total of 4 different pre-calculated datasets that can be loaded directly with the rstoolbox; those are cath, scop,
scop2 and chain (for PDB chains). For this datasets, homology (as provided by the PDB database) can be used to avoid over-represented
structures. The access to these datasets is provided through the load_refdata() function:
In [1]: import rstoolbox as rs
...: import pandas as pd
...: import matplotlib.pyplot as plt
...: import seaborn as sns
...: plt.rcParams['svg.fonttype'] = 'none' # When plt.savefig to 'svg', text is still text object
...: sns.set_style('whitegrid')
...: pd.set_option('display.width', 1000)
...: scop2 = rs.utils.load_refdata('scop2')
...: scop2_70 = rs.utils.load_refdata('scop2', 70) # 70% homology filter
...: scop2_70.drop(columns=['sequence_$', 'structure_$', 'psi_$', 'phi_$', 'node_name']).head(5)
...:
Out[1]:
scop_id serial pdb chain begin end selectors leaf domain_id score hbond_sr_bb hbond_lr_bb hbond_bb_sc hbond_sc dslf_fa13 omega p_aa_pp yhh_planarity ref rama_prepro BUNS CYDentropy avdegree beta breaks cavity charge exposed_hyd helix long_apolar long_polar pack psipred radius length node_id
0 8003767 1 2UWE I 1 99 (I:1-99,) True 2UWEI80037671 -237.172 -8.136 -42.638 -17.730 -7.007 -1.163 9.673 -23.884 0.026 8.637 -8.386 2.0 7.735 10.010 6.0 0.0 10.279 -2.0 932.371 0.0 6.0 6.0 0.661 0.727 14.066 99 [2000051, 6001103, 5000763, 4000202, 3000071]
1 8003798 1 2OAN C 4 371 (C:4-371,) True 2OANC80037981 -898.795 -139.883 -78.118 -37.999 -39.343 0.000 33.758 -72.597 0.036 121.341 2.231 31.0 0.000 11.330 13.0 1.0 294.330 -12.0 2812.701 15.0 7.0 6.0 0.657 0.675 21.333 357 [3000092, 6001267, 5000898, 4000313]
2 8003706 1 2UUB M 2 125 (M:2-125,) True 2UUBM80037061 -74.236 -56.345 -4.286 -7.571 -5.800 0.000 6.343 -22.039 0.001 43.878 10.694 17.0 0.000 9.516 0.0 0.0 41.054 19.0 1472.719 6.0 6.0 8.0 0.631 0.758 21.976 124 [6001039, 5000712, 4000260]
3 8017549 1 1FQB A 2 370 (A:2-370,) True 1FQBA80175491 -957.723 -144.574 -69.207 -53.420 -29.550 0.000 15.504 -71.976 0.045 83.748 4.174 42.0 0.000 11.715 12.0 0.0 432.225 -9.0 2157.583 16.0 8.0 5.0 0.671 0.702 21.546 369 [6002815, 5001965]
4 8004502 1 1W8M A 2 164 (A:2-164,) True 1W8MA80045021 -461.079 -36.431 -56.442 -37.828 -15.055 0.000 17.157 -58.100 0.004 57.392 -3.910 17.0 0.000 11.294 8.0 0.0 38.591 2.0 1157.008 2.0 7.0 10.0 0.786 0.687 14.256 163 [6001369, 5000991, 4000390, 3000168, 2000164]
These sets are only provided as help, but the user can create its own sets, as long as they are loaded as a DataFrame.
Cleaning your own background sets¶
In case one has its own background reference dataset, rstoolbox offers the option to retrieve homology clusters precalculated
from PDB with the make_redundancy_table() function. By default, this function will call on the PDB’s ftp to download those
homology cluster files, but with precalculated==True it will directly provide a quicker access to the table available in the
library (as the first option is relatively time consuming).
The generated table can be used on your reference set as long as a pdb and chain column exists.
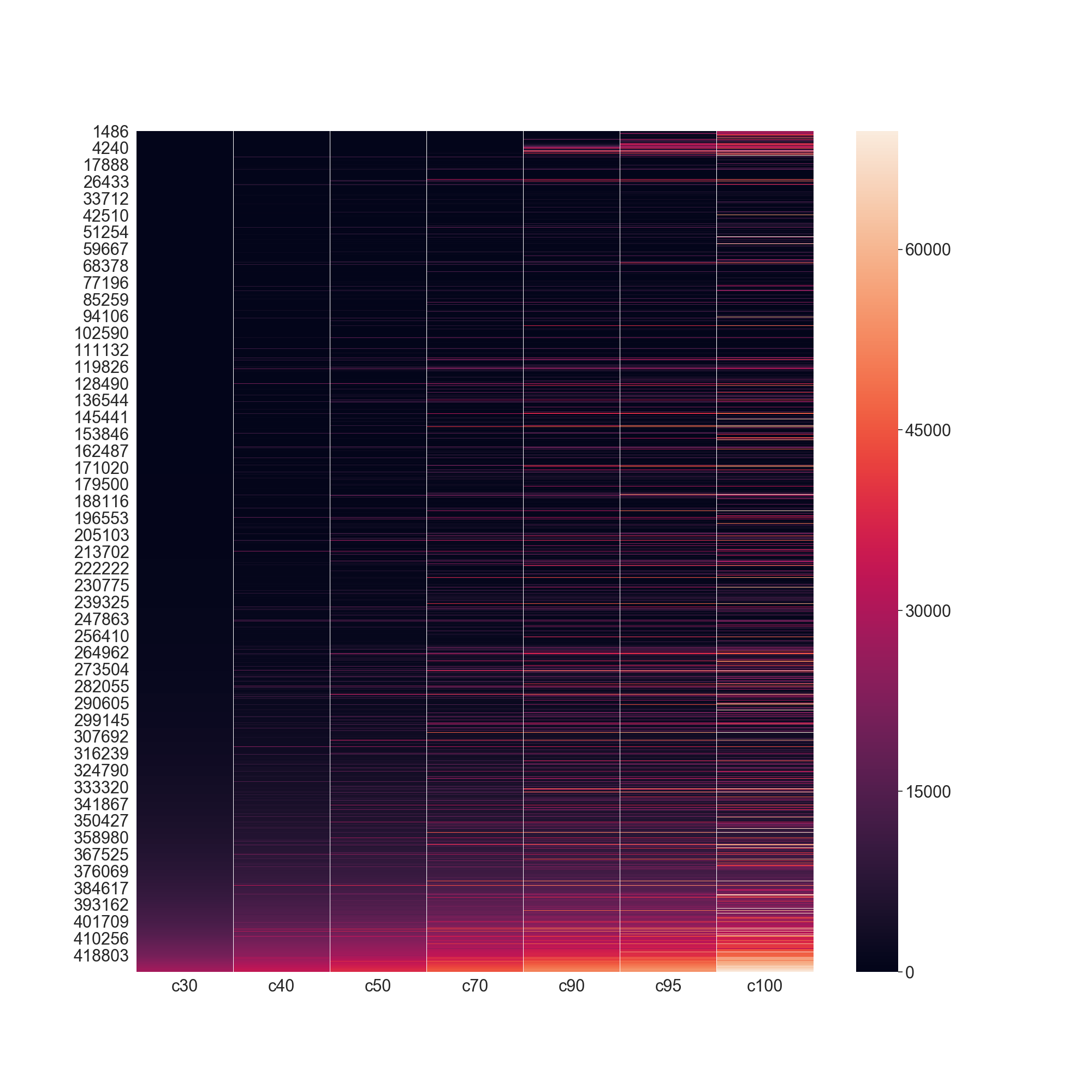
The following example shows now this table can be applied in what would be the equivalent of rs.utils.load_refdata('scop2', 30)
and creates a visual representation of the clusters.
In [2]: hmdf = rs.utils.make_redundancy_table(precalculated=True)
...: scop2_30 = scop2.merge(hmdf, on=['pdb', 'chain'])
...: scop2_30 = scop2_30.sort_values('score').groupby('c30').first().reset_index()
...: fig = plt.figure(figsize=(20, 20))
...: ax = plt.subplot2grid((1, 1), (0, 0), fig=fig)
...: _ = sns.heatmap(hmdf.drop(columns=['pdb', 'chain']).sort_values(['c30', 'c40', 'c50', 'c70', 'c90', 'c95', 'c100']), ax=ax)
...:
In [3]: plt.show()
Distribution in context¶
Let’s assume the following population dataset:
In [4]: rules = {'scores_ignore': ['fa_*', 'niccd_*', 'hbond_*', 'lk_ball_wtd', 'pro_close', 'dslf_fa13', 'C_ni_rmsd_threshold',
...: 'omega', 'p_aa_pp', 'yhh_planarity', 'ref', 'rama_prepro', 'time'],
...: 'sequence': 'C',
...: 'labels': ['MOTIF', 'SSE03', 'SSE05']}
...: df = rs.io.parse_rosetta_file('../rstoolbox/tests/data/input_ssebig.minisilent.gz', rules)
...: df.head(3)
...:
Out[4]:
score ALIGNRMSD BUNS COMPRRMSD C_ni_mtcontacts C_ni_rmsd C_ni_trials MOTIFRMSD cav_vol driftRMSD finalRMSD packstat C_ni_rmsd_type description lbl_MOTIF lbl_SSE03 lbl_SSE05 sequence_C
0 -64.070 0.608 12.0 7.585 4.0 3.301 1.0 0.957 66.602 0.083 3.323 0.544 no_motif nubinitio_wauto_18326_2pw9C_0001_0001 [C] [C] [C] TTWIKFFAGGTLVEEFEYSSVNWEEIEKRAWKKLGRWKKAEEGDLMIVYPDGKVVSWA
1 -70.981 0.639 12.0 2.410 8.0 1.423 1.0 0.737 0.000 0.094 1.395 0.552 no_motif nubinitio_wauto_18326_2pw9C_0002_0001 [C] [C] [C] NTWSTNILNGHPKITLLVEERGAEEIHLEWLKKQGLRKKAEENVYTTKLPNGAVKVYG
2 -43.863 0.480 8.0 4.279 6.0 2.110 1.0 0.819 93.641 0.110 2.106 0.575 no_motif nubinitio_wauto_18326_2pw9C_0003_0001 [C] [C] [C] PRWFIAMGDGVIWEIVLGSEQNLEEIAKKGLKRRGLYKKAEESIYTIIYPDGIAHTFG
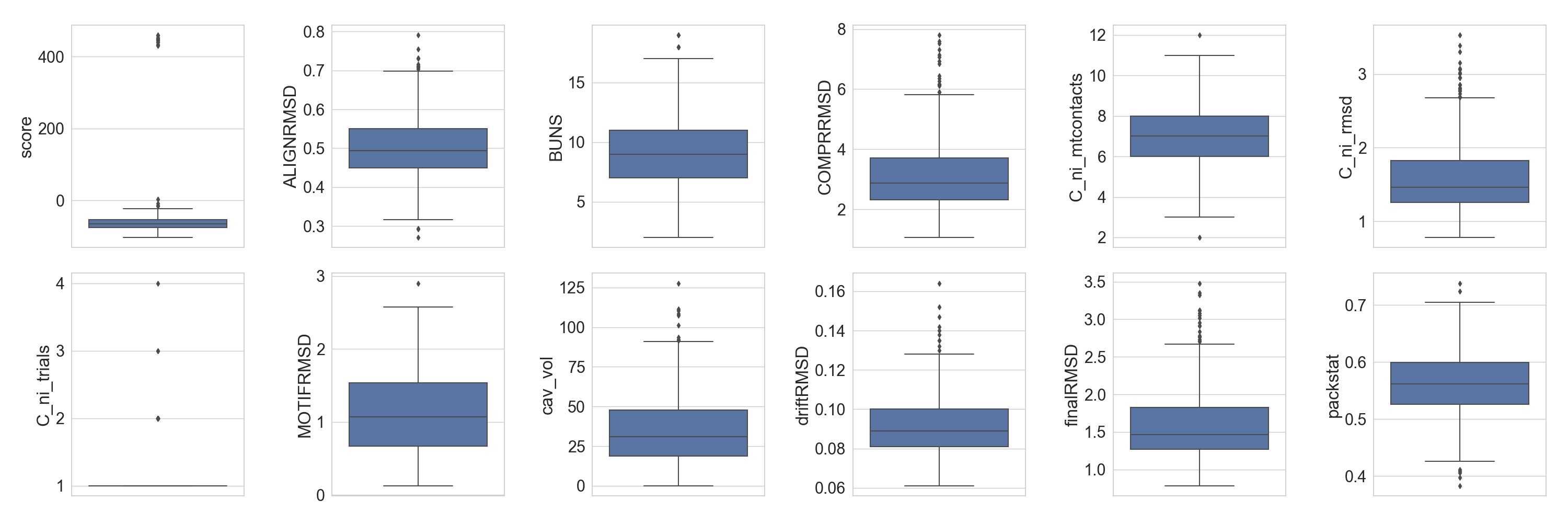
As previously demonstrated in the sequence analysis tutorial, we can preview the distribution of different scores of the design population:
In [5]: fig = plt.figure(figsize=(30, 10))
...: grid = [2, 6]
...: axes = rs.plot.multiple_distributions( df, fig, grid )
...: plt.tight_layout()
...:
In [6]: plt.show()
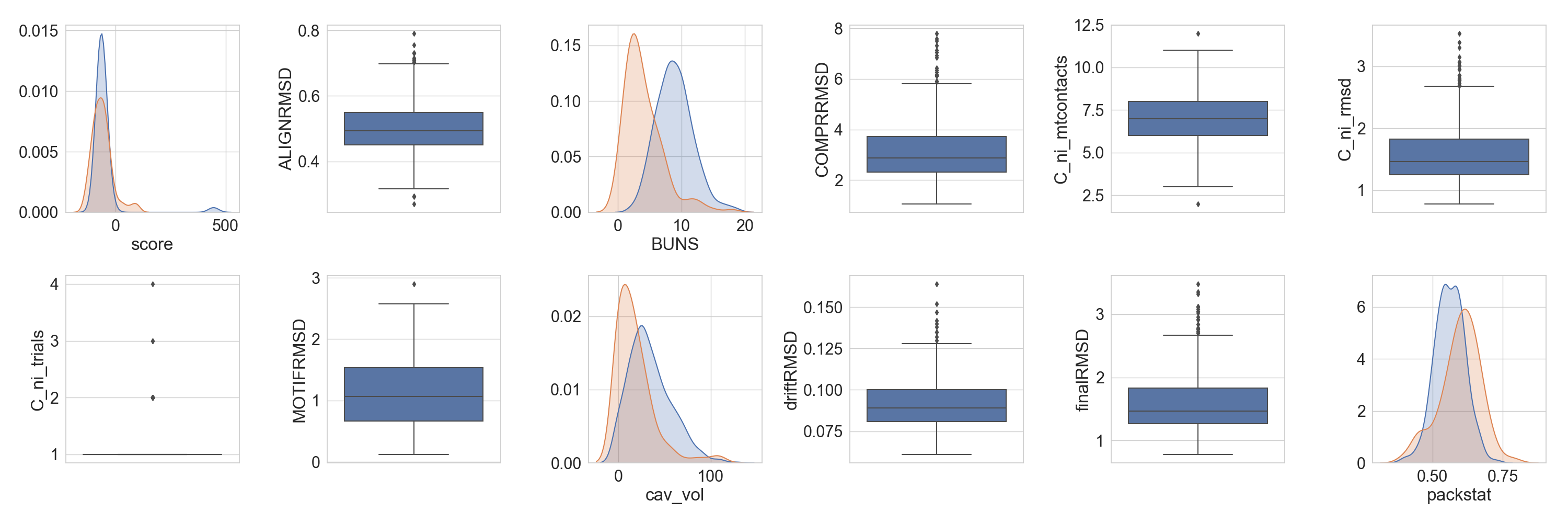
But we can also see the population behave against other protein of a similar size.
Note
The function will pick up that columns with the same name are the same, for others that should be the same but are called
differently, a translation dictionary can be provided, as is the case in this example with cavity volume and packstat
data.
In [7]: slength = len(df.iloc[0].get_sequence('C'))
...: refdf = scop2[(scop2['length'] >= slength - 5) &
...: (scop2['length'] <= slength + 5) &
...: (scop2['score'] <= 100)]
...: fig = plt.figure(figsize=(30, 10))
...: axs = rs.plot.multiple_distributions(df, fig, grid, refdata=refdf, violins=False,
...: ref_equivalences={'cavity': 'cav_vol', 'pack': 'packstat'})
...: plt.tight_layout()
...:
In [8]: plt.show()
Selections in context¶
Contrary to the previous example, in which full distributions where compared, another useful tool is the ability to place selected structures in a given context. This can have two main uses: (1) see the position of selected decoys in a given distribution or (2) see the quality of putative template scaffolds before working with them.
In [9]: pickdf = df.sample(10)
...: fig = plt.figure(figsize=(20, 5))
...: axs = rs.plot.plot_in_context(pickdf, fig, (1, 4), refdata=refdf, values= ['score', 'packstat', 'cav_vol', 'BUNS'],
...: ref_equivalences={'cavity': 'cav_vol', 'pack': 'packstat'})
...: plt.tight_layout()
...:
In [10]: plt.show()

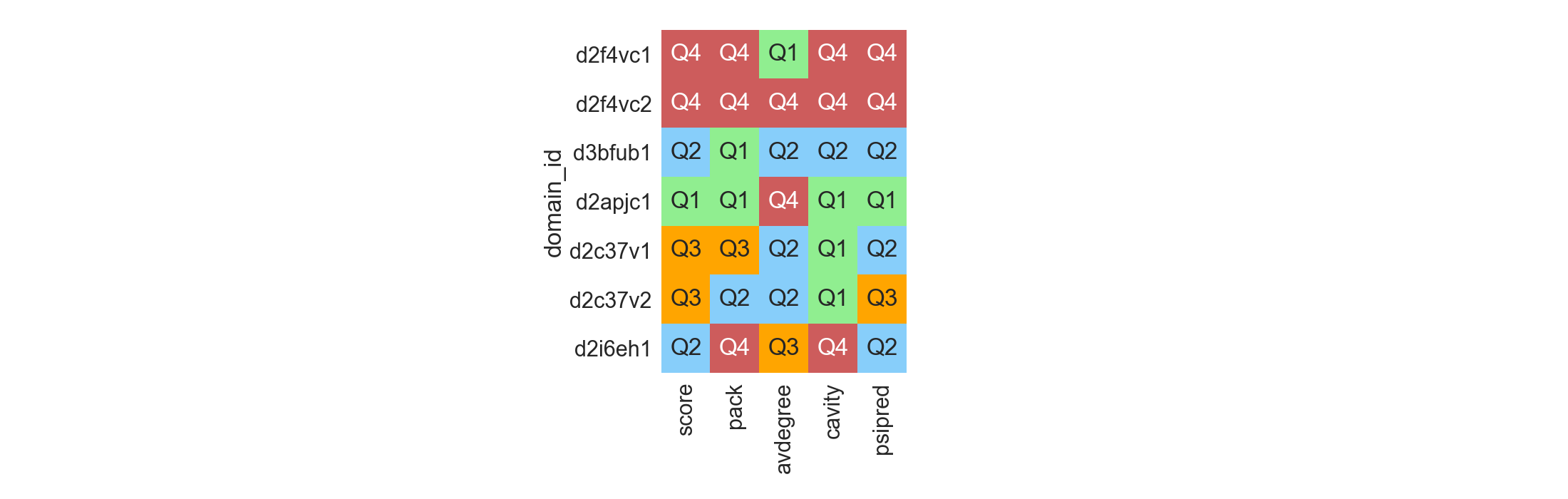
Each selection in its context¶
Multiple selected decoys or scaffolds originated from different sources might not be all comparable under the same set. The most simple case would be when those structures do not have a similar length, which does affect Rosetta scores. In those scenarios, it is possible to see, for each decoy of interest, how well it compares to its reference dataset, and rank them according to which quantile of the distribution they belong to:
In [11]: df = rs.utils.load_refdata('scop')
....: qr = pd.DataFrame([['2F4V', 'C'], ['3BFU', 'B'], ['2APJ', 'C'],
....: ['2C37', 'V'], ['2I6E', 'H']], columns=['pdb', 'chain'])
....: qr = qr.merge(df, on=['pdb', 'chain'])
....: refs = []
....: for i, t in qr.iterrows():
....: refs.append(df[(df['length'] >= (t['length'] - 5)) &
....: (df['length'] <= (t['length'] + 5))])
....: fig = plt.figure(figsize=(22, 7))
....: rs.plot.distribution_quality(df=qr, refdata=refs,
....: values=['score', 'pack', 'avdegree', 'cavity', 'psipred'],
....: ascending=[True, False, True, True, False],
....: names=['domain_id'], fig=fig)
....: plt.tight_layout()
....:
In [12]: plt.show()