Structural Analysis¶
Structural data can be obtained in multiple ways, and it should be relatively easy to implement a DSSP or PSIPRED parser to include that data into the decoy population.
In line of a Rosetta pipeline, the easiest way is to include the WriteSSEMover into a RosettaScript. This could even be called from inside the library as explained in Sequence Analysis.
Loading a Reference¶
As in Sequence Analysis, we will need to load a reference with get_sequence_and_structure().
Note
Through all the process several times the chainID of the decoy of interest will be called. This is due to the fact that the library can manipulate
decoys with multiple chains (designed or not), and, thus, analysis must be called upon the sequences of interest.
In [1]: import rstoolbox as rs
...: import pandas as pd
...: import matplotlib.pyplot as plt
...: import seaborn as sns
...: plt.rcParams['svg.fonttype'] = 'none' # When plt.savefig to 'svg', text is still text object
...: sns.set_style('whitegrid')
...: pd.set_option('display.width', 1000)
...: pd.set_option('display.max_columns', 500)
...: pd.set_option("display.max_seq_items", 3)
...: baseline = rs.io.get_sequence_and_structure('../rstoolbox/tests/data/2pw9C.pdb')
...: baseline.get_structure('C')
...:
Out[1]:
0 LEEEEEEELLEEEEEEEELLLLHHHHHHHHHHHHLLLLLLLLLLLEEEELLLEEEELL
Name: structure_C, dtype: object
Loading the Design Data¶
Again, we are mimicking Sequence Analysis, but in this case we load the structural data of the designs along with their dihedrals.
In [2]: rules = {'scores_ignore': ['fa_*', 'niccd_*', 'hbond_*', 'lk_ball_wtd', 'pro_close', 'dslf_fa13', 'C_ni_rmsd_threshold',
...: 'omega', 'p_aa_pp', 'yhh_planarity', 'ref', 'rama_prepro', 'time'],
...: 'sequence': 'C', 'structure': 'C', 'dihedrals': 'C',
...: 'labels': ['MOTIF', 'SSE03', 'SSE05']}
...: df = rs.io.parse_rosetta_file('../rstoolbox/tests/data/input_ssebig.minisilent.gz', rules)
...: df.add_reference_structure('C', baseline.get_structure('C'))
...: df.add_reference_shift('C', 32)
...: df.head(3)
...:
Out[2]:
score ALIGNRMSD BUNS COMPRRMSD C_ni_mtcontacts C_ni_rmsd C_ni_trials MOTIFRMSD cav_vol driftRMSD finalRMSD packstat C_ni_rmsd_type description lbl_MOTIF lbl_SSE03 lbl_SSE05 sequence_C structure_C phi_C psi_C
0 -64.070 0.608 12.0 7.585 4.0 3.301 1.0 0.957 66.602 0.083 3.323 0.544 no_motif nubinitio_wauto_18326_2pw9C_0001_0001 [C] [C] [C] TTWIKFFAGGTLVEEFEYSSVNWEEIEKRAWKKLGRWKKAEEGDLMIVYPDGKVVSWA LEEEEEEELLLEEEEEEELLLLHHHHHHHHHHHHLLLLLLLLLLLEEEELLLEEEELL [0.0, -143.01, -92.3838, ...] [163.023, 142.263, 136.923, ...]
1 -70.981 0.639 12.0 2.410 8.0 1.423 1.0 0.737 0.000 0.094 1.395 0.552 no_motif nubinitio_wauto_18326_2pw9C_0002_0001 [C] [C] [C] NTWSTNILNGHPKITLLVEERGAEEIHLEWLKKQGLRKKAEENVYTTKLPNGAVKVYG LEEEEEEELLEEEEEEEELLLLHHHHHHHHHHHLLLLLLLLLLLEEEELLLLLEEEEL [0.0, -144.628, -82.9913, ...] [109.954, 146.837, 129.948, ...]
2 -43.863 0.480 8.0 4.279 6.0 2.110 1.0 0.819 93.641 0.110 2.106 0.575 no_motif nubinitio_wauto_18326_2pw9C_0003_0001 [C] [C] [C] PRWFIAMGDGVIWEIVLGSEQNLEEIAKKGLKRRGLYKKAEESIYTIIYPDGIAHTFG LEEEEEEELLEEEEEEEELLLLHHHHHHHHHHHHLLLLLLLLLLEEEEELLLEEEEEL [0.0, -143.373, -78.9875, ...] [141.285, 139.737, 129.8, ...]
Select by Structural Properties¶
One of the most obvious assessments is the calculation of percentages of secondary structure. We can even use that to pre-select decoys. For example,
let’s assume that we want to make sure that strand 5 (SSE05) from the selected labels is mostly still a strand after our design protocol:
In [3]: sse05 = df.get_label('SSE05', 'C').values[0]
...: df = rs.analysis.secondary_structure_percentage(df, 'C', sse05)
...: dfsse = df[df['structure_C_E'] > 0.8]
...: dfsse.head(3)
...:
Out[3]:
score ALIGNRMSD BUNS COMPRRMSD C_ni_mtcontacts C_ni_rmsd C_ni_trials MOTIFRMSD cav_vol driftRMSD finalRMSD packstat C_ni_rmsd_type description lbl_MOTIF lbl_SSE03 lbl_SSE05 sequence_C structure_C phi_C psi_C structure_C_H structure_C_E structure_C_L
2 -43.863 0.480 8.0 4.279 6.0 2.110 1.0 0.819 93.641 0.110 2.106 0.575 no_motif nubinitio_wauto_18326_2pw9C_0003_0001 [C] [C] [C] PRWFIAMGDGVIWEIVLGSEQNLEEIAKKGLKRRGLYKKAEESIYTIIYPDGIAHTFG LEEEEEEELLEEEEEEEELLLLHHHHHHHHHHHHLLLLLLLLLLEEEEELLLEEEEEL [0.0, -143.373, -78.9875, ...] [141.285, 139.737, 129.8, ...] 0.0 0.833333 0.166667
3 -75.847 0.454 7.0 3.112 8.0 1.746 1.0 1.629 39.232 0.083 1.734 0.644 no_motif nubinitio_wauto_18326_2pw9C_0004_0001 [C] [C] [C] PYEWVFIINGVPQTTWNHPPTKMEELEKFARKKGGSSKKAEEGKFAIIIWKGYFIVSD LLLEEEEELLEEEEEELLLLLLHHHHHHHHHHHHLLLLLLLLLLEEEEELLLEEEEEL [0.0, -130.948, -160.432, ...] [141.392, 174.651, 115.305, ...] 0.0 0.833333 0.166667
6 -47.169 0.430 6.0 3.313 6.0 2.050 1.0 0.641 0.000 0.112 1.975 0.551 no_motif nubinitio_wauto_18326_2pw9C_0007_0001 [C] [C] [C] PREWEGRYNGVPRGREWALPTNLEELMKEMRKRAGGYKKAEEGIYATIFPNGVILVRG LEEEEEEELLEEEEEEEELLLLHHHHHHHHHHHHLLLLLLLLLLEEEEELLLEEEEEL [0.0, -133.286, -93.7573, ...] [142.271, 153.259, 120.778, ...] 0.0 0.833333 0.166667
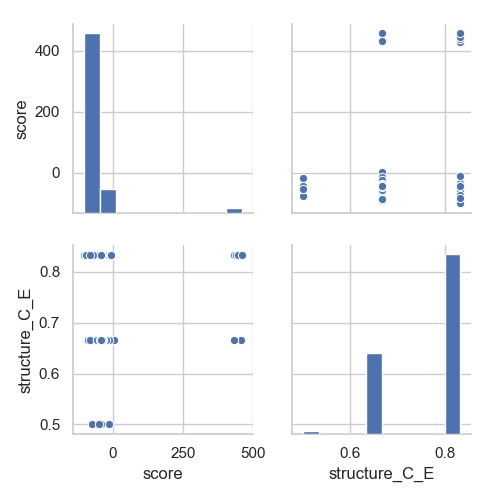
Or that we want to check if there is a correlation between the final score of our decoys and the level of conservation of strand 5 (which does not seem to be the case):
In [4]: grid = sns.pairplot(df[['score', 'structure_C_E']])
In [5]: plt.show()
Visualise Structural Data¶
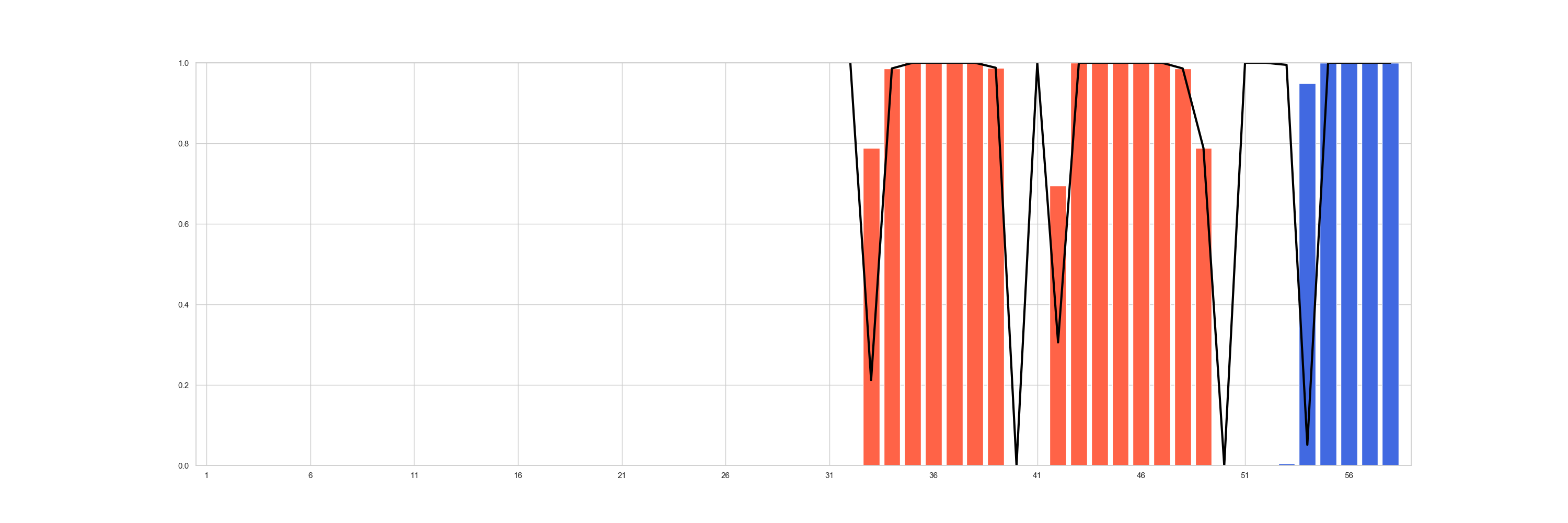
The most global view, would be to see for all the population how often the expected secondary structure is retrieved with
positional_structural_similarity_plot().
In [6]: fig = plt.figure(figsize=(30, 10))
...: ax = plt.subplot2grid((1, 1), (0, 0))
...: sseid1 = rs.analysis.positional_structural_count(df, 'C')
...: sseid2 = rs.analysis.positional_structural_identity(df, 'C')
...: rs.plot.positional_structural_similarity_plot(pd.concat([sseid1, sseid2], axis=1), ax)
...:
In [7]: plt.show()
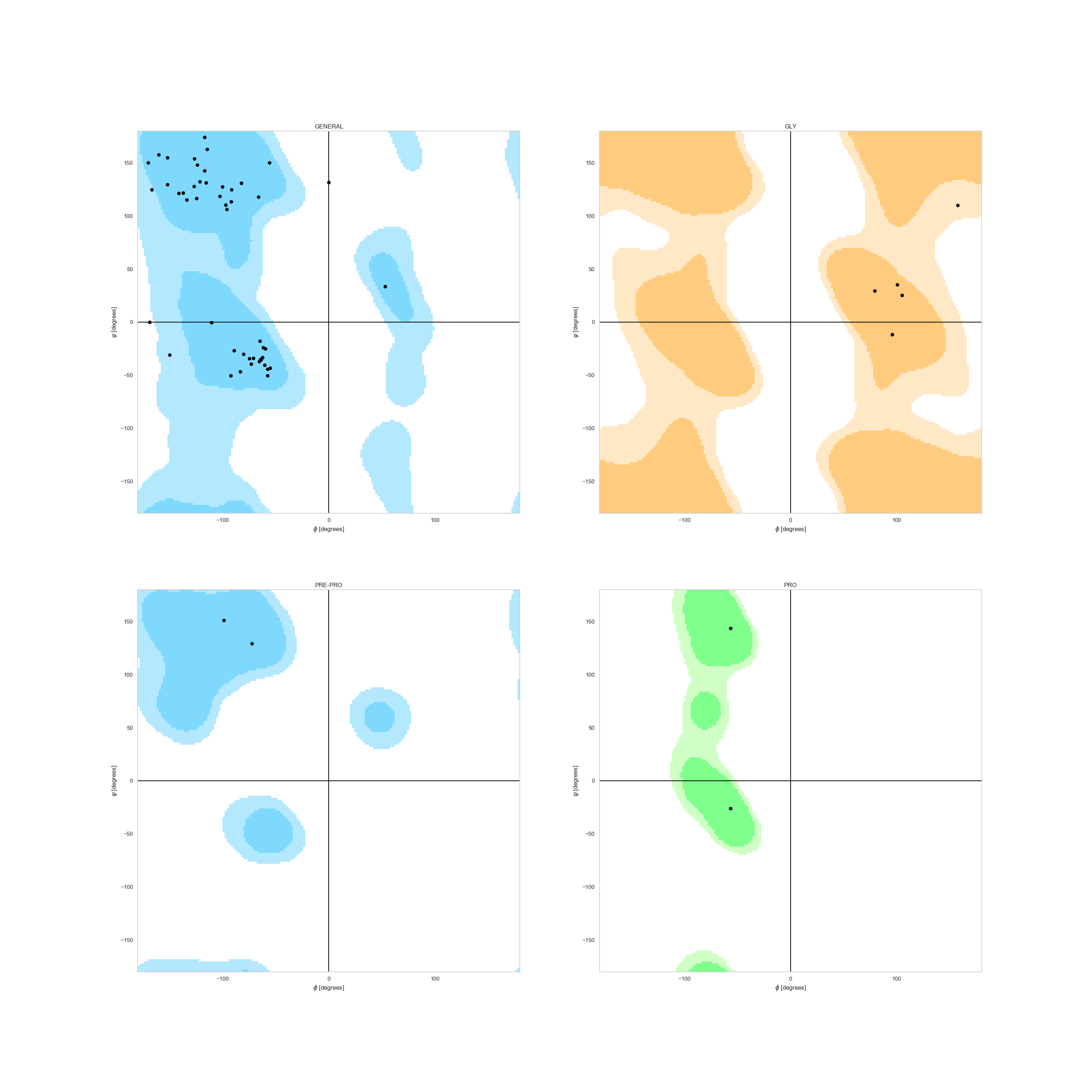
In case of a single decoy, and thanks to our request for dihedrals in the parsing rules, we can check whether or not
a the decoy’s Ramachandran Plot makes sense. We can observe it, for example, for the best
scored decoy under the plot_ramachandran() function.
Note
Dihedral angles are also obtained through WriteSSEMover,
but they can easily be added by parsing them into a list and adding them to the DesignFrame with the column name psi_<seqID> and phi_<seqID>.
In [8]: fig = plt.figure(figsize=(30, 30))
...: _ = rs.plot.plot_ramachandran(df.sort_values('score').iloc[0], 'C', fig)
...:
In [9]: plt.show()
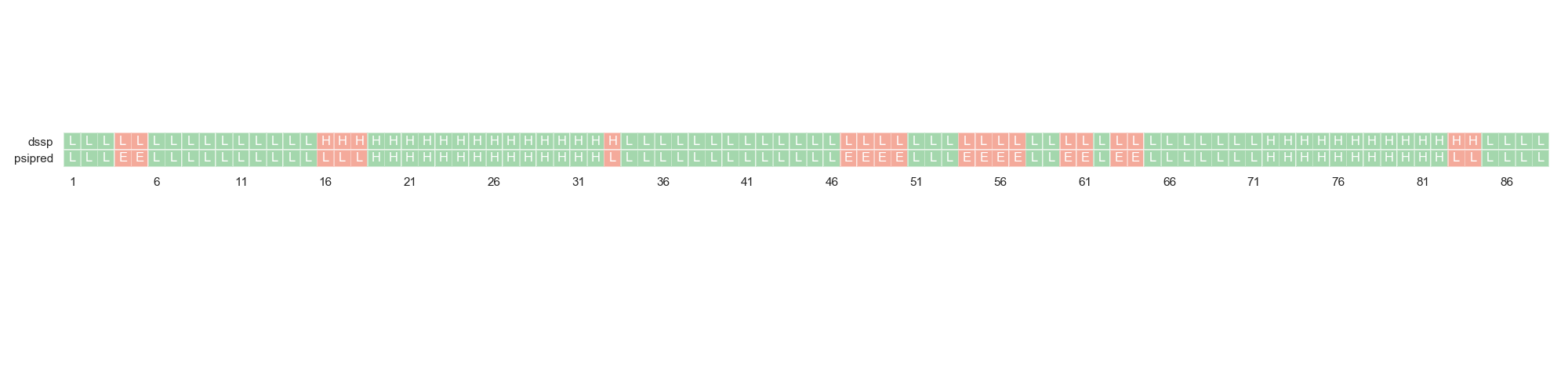
Finally, one can compare the actual DSSP assignation against PSIPRED’s sequence-based secondary structure prediction. This can give an idea of the feasibility of the sequence to actually fold in the expected conformation.
For this, we are going to load a different dataset in which this data is present.
In [10]: fig = plt.figure(figsize=(20, 5))
....: rules = {"scores": ["score"], "psipred": "*", "structure": "*", "dihedrals": "*" }
....: df2 = rs.io.parse_rosetta_file("../rstoolbox/tests/data/input_3ssepred.minisilent.gz", rules)
....: ax = plt.subplot2grid((1, 1), (0, 0))
....: rs.plot.plot_dssp_vs_psipred(df2.iloc[0], "A", ax)
....: plt.tight_layout()
....: fig.subplots_adjust(top=1.2)
....:
In [11]: plt.show()
In [12]: plt.close('all')